Что происходит в геномах бактерий
В разных линиях и штаммах бактерий могут независимо друг от друга возникать различные хромосомные перестройки. Это, например, инверсия (поворот участка хромосомы на 180 градусов), делеция (потеря участка хромосомы), дупликация (удвоение участка хромосомы) и так далее. В результате эволюции бактерии постоянно меняются: в частности, они могут адаптироваться к внешним условиям, а также конкурировать друг с другом и избегать иммунного ответа организма-хозяина.
Подобные перестройки могут стать причиной вирулентности (способности штамма микроорганизма или вируса заражать организм), устойчивости к антибиотикам или антигенной изменчивости.
Зачастую, чтобы выявить все эти события и проанализировать их, требуется кропотливая лабораторная проверка, которая занимает много времени.
Как быстро проанализировать изменения в бактериологическом геноме
Ученые ИТМО разработали метод, который назвали PaReBrick (PArallel REarrangements and BReaks identification toolkit). С его помощью можно быстро вычленить и проанализировать параллельные адаптации в популяциях бактерий.
«Допустим, мы берем несколько организмов и находим у них общие блоки — так называемые блоки синтении, консервативные геномные последовательности. Эти блоки довольно длинные и в процессе эволюции могут меняться между собой местами. В геноме может произойти вставка или, например, может развернуться определенный сегмент. В итоге у нас получается разная последовательность блоков для каждого организма, на которую пока тяжело смотреть систематически. Мы же разработали метод, позволяющий упростить этот процесс», — поясняет соавтор работы, аспирант факультета информационных технологий и программирования Университета ИТМО Алексей Забелкин.
Алексей Забелкин. Фото из личного архива
Создавая метод, исследователи опирались на работу ученых ИППИ РАН, посвященную микроэволюции различных видов стрептококка.
«Коллеги обнаружили очень интересную вещь. Есть патогенные штаммы стрептококков, вызывающие пневмонию. У этих бактерий существуют два гена — PhtD и PhtB, кодирующие белки поверхностной мембраны. Эти белки — антигены: маркер, по которому иммунная система человека распознает и уничтожает клетки этого патогена. Ученые показали, что у некоторых штаммов происходит рекомбинация по повторам внутри этих генов. Это не случайность, а эволюционный механизм — поверхностные белки, с которыми потенциально может связываться наша иммунная система. И изменения в них — способ ухода от иммунного ответа. Что самое интересное, один из этих белков был отобран как таргет для разработки новой пневмококковой вакцины, которая уже прошла несколько стадий клинических испытаний, однако рекомбинантные варианты белков не были учтены», — рассказывает Алексей Забелкин.
Ученые ИТМО решили формализовать задачу анализа бактериальных геномов и разработать инструмент, который позволяет выявлять параллельные адаптации, соединив методы сравнительной геномики, филогенетики и алгоритмы обработки графов. Он использует филогенетическое дерево бактериальных штаммов для анализа расположения разных блоков, а затем автоматически находит параллельные перестройки в геномах.
Анализ генома бактерии Streptococcus pyogenes. Иллюстрация из статьи в журнале Bioinformatics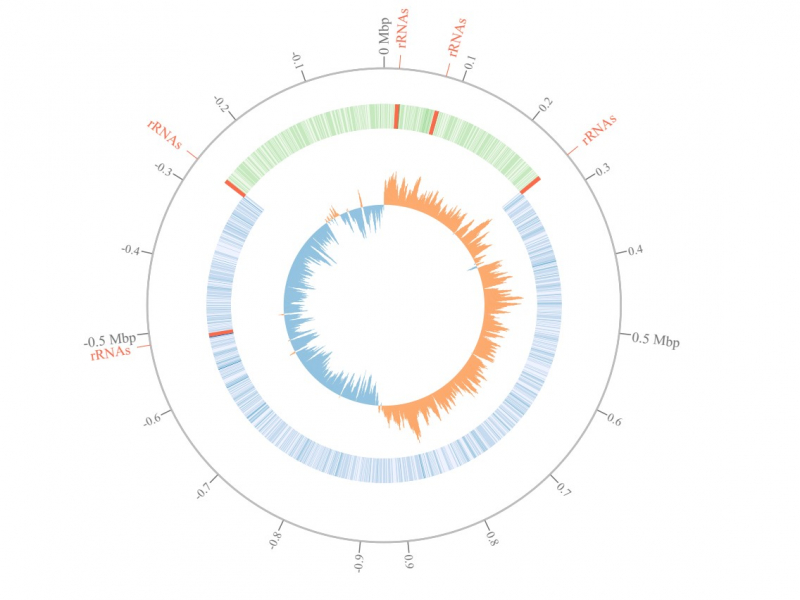
Однако изменений в бактериальном геноме довольно много, и следующей задачей было отыскать именно те, что потенциально могут быть интересны для дальнейшего исследования. Чтобы выявить их, ученые предложили метрику для оценки степени параллельности эволюционного события и ранжирование по ней.
Инструмент PaReBrick, разработанный исследователями, позволяет анализировать параллельные инверсии, вставки и потери у близкородственных бактерий и вычленять события, ответственные за параллельную адаптацию и изменение фенотипа. Иллюстрируя эффективность PaReBrick, ученые проанализировали датасеты из порядка 200 штаммов стрептококка и 150 штаммов патогенных кишечных палочек — алгоритм обработал эти данные всего за минуту. В ходе этого анализа были подтверждены ранее известные перестройки, а также найдены другие примеры параллельной адаптации. Например, инверсия по различным копиям рибосомального оперона у пиогенных стрептококков и параллельное приобретение патогенности у разных кишечных палочек.
Рибосомальный оперон — «Соседи». Иллюстрация из презентации разработки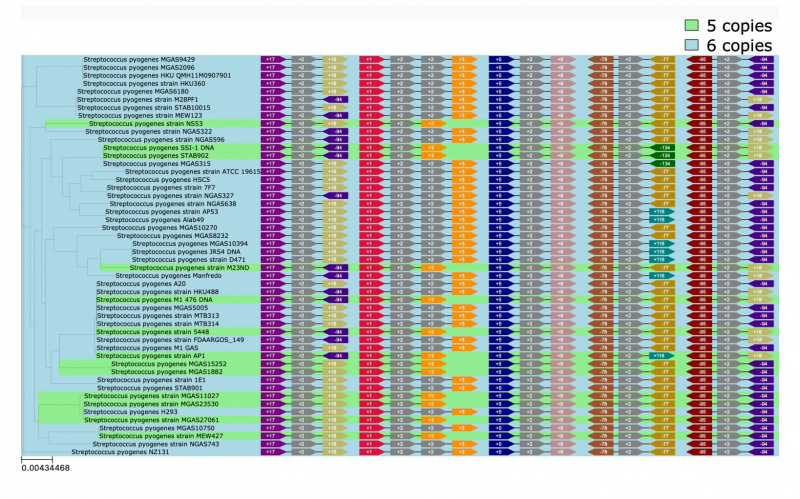
Где можно использовать метод
Разработанный метод пригодится для анализа и сравнения геномов любых бактерий:
«Обычно биологи находят подобную информацию в пробирке: ученый увидел изменения в определенном штамме, потом описал их. А именно систематический анализ большого количества штаммов с точки зрения эволюции последовательностей и с наложением этой информации на филогенетическое дерево до нас практически никто не делал», — заключает Алексей Забелкин.
Метод может применяться в различных фундаментальных исследованиях эволюционной и системной биологии. На практике он будет полезен для изучения медицински значимых штаммов, патогенов животных и растений, симбионтов сельскохозяйственных культур. Полученные результаты поспособствуют эффективной разработке тест-систем, вакцин и лекарств, а также могут быть использованы в области генной инженерии.
Подробнее о разработке: Alexey Zabelkin, Yulia Yakovleva, Olga Bochkareva, Nikita Alexeev, PaReBrick: PArallel REarrangements and BReaks identification toolkit (Bioinformatics, 2021).