What goes on in bacterial genomes
Various chromosome alterations can appear in different lineages and strains of bacteria independently. For example, there’s inversion (180-degree rotation of a chromosomal segment), deletion (loss of a chromosomal segment), duplication (repetition of a chromosomal segment), and so on. As a result of evolution, bacteria constantly change: they can adapt to external conditions, compete with each other, and avoid the immune response of their host.
Such alterations can lead to virulence – the ability of a strain of a microorganism or virus to infect the body, as well as be resistant to antibiotics or antigenic variation. Often, scientists need to conduct a thorough and time-demanding lab study to detect and analyze these events.
Quick analysis
The PaReBrick (PArallel REarrangements and BReaks identification toolkit) algorithm developed by scientists from ITMO University in collaboration with the Institute of Science and Technology Austria (IST Austria) allows users to automatically identify and analyze parallel adaptation events in bacterial populations.
“Let’s say we’re studying several organisms and detect common synteny blocks – conserved genome sequences. These blocks are quite long and might switch places as they evolve. A segment might be inserted or rotated. As a result, we end up with a different sequence of blocks for each organism and it’s hard to look at that systematically. We have developed a method that helps make this process easier,” says Alexey Zabelkin, a PhD student at ITMO University and co-author of this paper.

Alexey Zabelkin
The project was inspired by the work of fellow researchers from the Institute for Information Transmission Problems of the Russian Academy of Sciences on micro-evolution of various Streptococcus species.
“Our colleagues have detected an interesting fact. There are pathogen strains of streptococci that cause pneumonia. These bacteria have two genes – PhtD and PhtB, which encode the proteins of a surface membrane. The proteins are antigens – markers thanks to which the immune system detects and destroys the pathogen. The scientists observed that some strains have recombination inside these genes. This isn’t a coincidence but an evolutionary mechanism. These are surface proteins with which our immune system can potentially connect. The alterations in them are a way to avoid the immune response. Interestingly enough, one of these proteins was chosen as a target in the development of a new pneumococcal vaccine, which has already undergone several stages of clinical testing. However, recombinations of the proteins weren’t taken into consideration,” says Alexey Zabelkin.
Scientists from ITMO decided to formalize the analysis of bacterial genomes and develop a tool for the detection of parallel adaptations that will combine the methods of comparative genomics, phylogenetics, and graph processing algorithms. This method uses the phylogenetic tree of bacterial strains to analyze the placement of various blocks and then looks for parallel alterations in genomes.
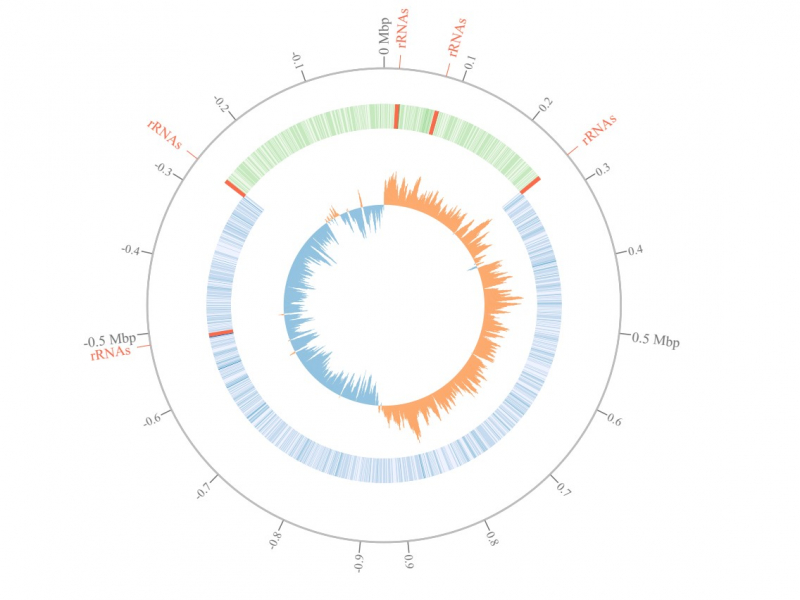
Analysis of bacterial genome of Streptococcus pyogenes. An illustration from the article in Bioinformatics journal.
However, there are quite a lot of changes in the bacterial genome, so the next task was to detect the relevant ones. For that, the scientists came up with a metric for evaluation of how parallel an evolutionary event is that helps rank them.
The PaReBrick algorithm developed by the researchers allows them to automatically identify parallel adaptation events in bacterial populations and detect events responsible for parallel adaptation and changes in the phenotype. To prove the efficiency of PaReBrick, the scientists tested it on about 200 strains of Streptococcus and 150 strains of pathogenic Escherichia coli – the algorithm completed the processing in only one minute. During the analysis, previously known alterations were confirmed and new ones discovered. For example, inversion in different copies of a ribosomal operon in Streptococcus pyogenes and acquisition of new pathogenicity factors in Escherichia coli.
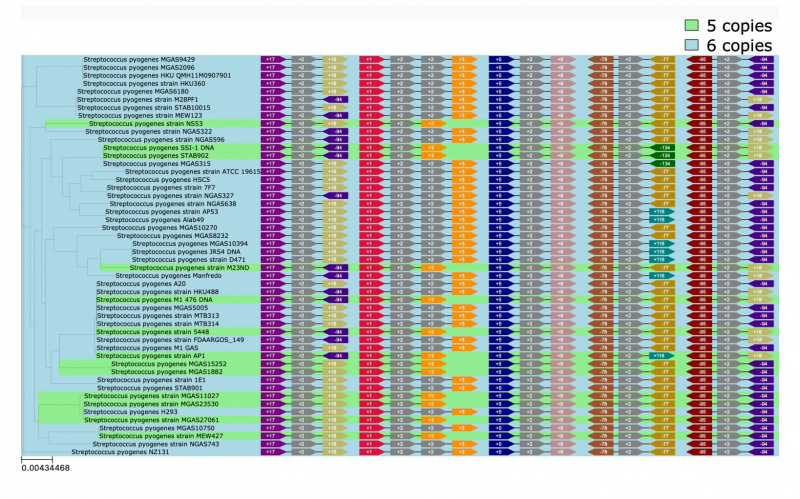
Ribosomal operon – "Neighbors". An illustration from the project's presentation.
Applications
The method can be used for the analysis of genomes of any bacteria.
“Usually biologists observe such phenomena in vitro: they detect alterations in a strain and describe them. But a systematic analysis of many strains from the point of view of the evolution of sequences and an application of this data in phylogenetic trees has barely been done before,” says Alexey Zabelkin.
The method can be applied in various fundamental research in the fields of evolutionary and systems biology. In practice, it will be useful for studying medically relevant strains, animal and plant pathogens, and symbionts of agricultural systems. The achieved results will help develop more efficient tests, vaccines, and drugs, as well as benefit genetic engineering.
Reference: Alexey Zabelkin, Yulia Yakovleva, Olga Bochkareva, Nikita Alexeev, PaReBrick: PArallel REarrangements and BReaks identification toolkit (Bioinformatics, 2021).