
Великий исход из Африки
Учёные провели полногеномный анализ исторических образцов леопардов, хранящихся в естественнонаучных музеях, и современной ДНК ныне живущих подвидов леопарда — представителей подвидов Африки и Азии. Главной целью исследования было изучить генетическую структуру мировой популяции леопарда, используя новые технологии и полногеномные данные.
Самым главным открытием для исследователей стало то, насколько сильны геномные различия между леопардами, живущими в Азии, и леопардами в Африке. Сами авторы приводят в пример различия между бурыми и белыми медведями в случае леопардов геномное расстояние даже больше. Причем, как удалось установить в ходе анализа, расхождение двух групп леопардов произошло около 500 – 600 тысяч лет назад — примерно в то же время, когда неандертальцы отделились от предков современных людей. Как считают ученые, миграция из Африки в Азию произошла один единственный раз — и это определило их сильнейшие геномные расхождения, которые только укреплялись последующие сотни тысяч лет.
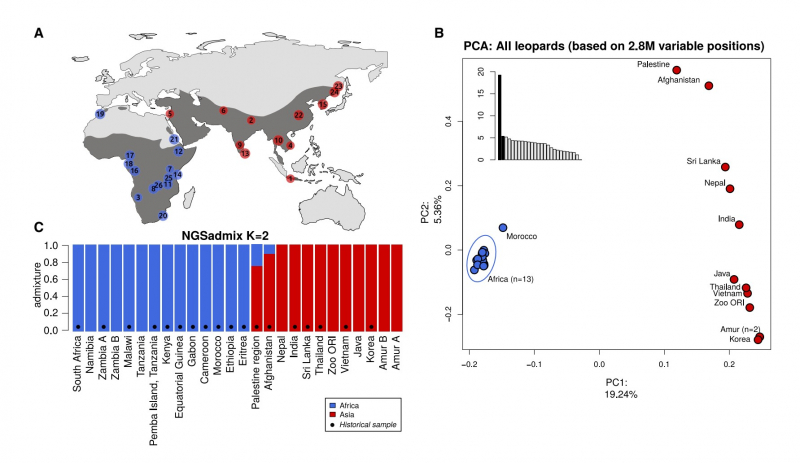
Распространение леопардов. Иллюстрация из статьи в Current Biology
Данное исследование проведено российскими исследователями из Университета ИТМО и Федерального Научного Центра Биоразнообразия Наземной Биоты Восточной Азии в составе международной команды ученых из Университета Ноттингем Трент, Кембриджского университета, Университета Лестера и Потсдамского Университета в Германии.
Работа длиной в 20 лет
Первое исследование ДНК современных леопардов проведено ровно 20 лет назад соавторами работы — Стивеном О'Брайеном, главным научным сотрудником лаборатории геномного разнообразия Университета ИТМО, и Ольгой Уфыркиной, старшим научным сотрудником ФНЦ Биоразнообразия ДВО РАН. Тогда им удалось проанализировать ДНК 77 леопардов из 13 географических регионов. Анализ этих образцов позволил выделить девять генетически различных подвидов. Как объясняет Стивен О'Брайен, с тех пор методы и технологии геномных исследований существенно продвинулись вперед и дали возможность провести более масштабный и глубокий анализ.
«В 2001 году мы опубликовали филогеографический анализ и исследование геномного разнообразия современных подвидов леопарда, в том числе и дальневосточного. Мы использовали методы, которые были доступны в то время: сравнение микросателлитной ДНК, филогенетический анализ митохондриальных последовательностей и другие. Уже тогда мы показали происхождение современных линий леопарда в Африке и его последующую миграцию в Азию. Мы обнаружили большее генетическое разнообразие африканских леопардов в сравнении с азиатскими и генетическое обеднение амурских леопардов, если сравнивать их с другими подвидами. Мы также выявили значительный генетический разрыв между африканскими и азиатскими видами, но ошиблись с его датированием: тогда мы предположили, что разделение произошло 170 000 – 300 000 лет назад. Сейчас с появлением новых методов и снижением в десятки раз стоимости полногеномного секвенирования мы с коллегами из Германии и Великобритании решили вернуться ко всем этим вопросам, что дало нам более глубокое понимание эволюции вида».
Генетическое истощение
Как известно, показателем успешного долгосрочного существования любого вида является достаточный уровень генетического разнообразия, или генетической изменчивости. Проведенное исследование показало, что азиатские леопарды сохраняют заметно меньшее генетическое разнообразие, чем африканские. Это объясняется единственным случаем миграции лишь небольшой части африканских леопардов в Азию, а также существованием больших географических барьеров между популяциями на азиатском континенте, что препятствует обмену генами между особями.
Среди азиатских подвидов, дальневосточным леопардам не повезло больше всего. Ученые подтвердили обнаруженный еще в ранних работах самый низкий уровень генетического разнообразия у этих леопардов среди всех изученных подвидов и показали конкретные признаки, свидетельствующие об инбридинге — близкородственных скрещиваниях внутри популяции.
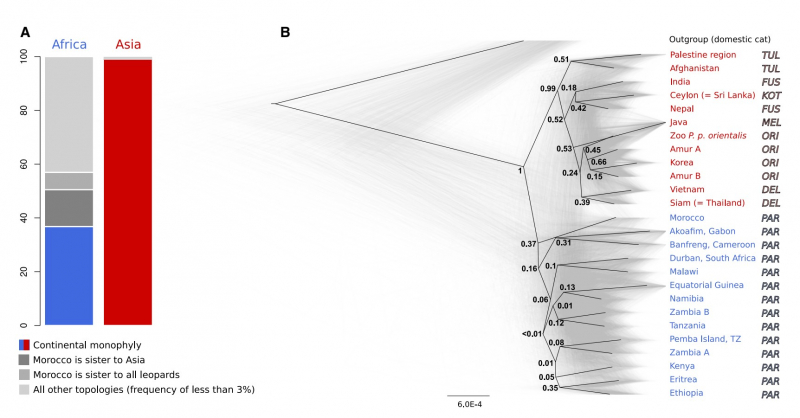
Анализ генома леопардов. Иллюстрация из статьи в Current Biology
Среди причин этого можно назвать крайний северный ареал обитания в природе, существенное сокращение численности в XX веке и существующую на протяжении очень долгого времени изолированность единственной популяции. Высокий уровень инбридинга ведет к истощению общего генетического материала и появлению вредных мутаций, которые подрывают жизнеспособность популяций в долгосрочном периоде.
Оздоровление популяции
Несмотря на то, что численность дальневосточных леопардов увеличилась за последние годы почти вдвое благодаря принятым природоохранным мерам и поддержке государства, генетическое разнообразие внутри популяции не может увеличиваться из-за отсутствия притока новых генов и подвид все еще балансирует на грани вымирания. Быстрое распространение нехарактерных для диких животных морфологических признаков — «белых лап» и укороченных хвостов — говорит о необходимости генетических исследований по поиску вредоносных генетических мутаций.
«Показанный на геномном уровне высокий уровень инбридинга свидетельствует о высокой уязвимости популяции и подтверждает необходимость реализации программы реинтродукции, которая была разработана специалистами еще 20 лет назад. В своих ранних работах мы показали, что амурские леопарды из зоопарков обладают бóльшим генетическим потенциалом, нежели природные особи, и при правильном подборе пар могут служить подходящим генетическим материалом для оздоровления природной популяции. Более того, создание второй резервной популяции в бывших местообитаниях леопарда послужило бы дополнительной гарантией сохранения грациозной кошки», — объясняет Ольга Уфыркина, старший научный сотрудник ФНЦ Биоразнообразия ДВО РАН.
Ученые считают, что дальнейшие исследования геномов дальневосточных леопардов помогут не только подобрать потенциально подходящих леопардов для выпуска их потомства в природу, но и создать программу генетического мониторинга для программы реинтродукции. Подобный проект разработан и успешно реализован во Флориде 15 лет назад — тогда удалось спасти популяцию флоридской пантеры благодаря выпуску в природный парк Флориды пантер из Техаса.
База данных ДНК всех существ
Исследование леопардов является частью поистине глобального проекта Genome 10K, который основан в 2009 году Дэвидом Хауслером, Оливером Райдером и Стивеном О'Брайеном. Цель проекта — собрать как можно больше данных о геномах ныне живущих животных. Такая «библиотека ДНК» станет базой для современных и будущих научных исследований, а также послужит для сохранения информации о вымирающих видах. Сейчас в научное сообщество проекта (G10K Consortium) входят 150 специалистов из 50 институтов по всему миру.
Флагманский проект — The Vertebrate Genomes Project — предполагает полное геномное секвенирование всех известных позвоночных (то есть 71 657 видов животных) в течение ближайших десяти лет. Обзор проекта и первые результаты работы были недавно опубликованы в журнале Nature.
«Когда мы только начали проект G10K, мы собрали небольшой круг полевых зоологов — вместе с биоинформатиками, чьи интересы лежат в области геномики. Мы поставили себе цель собрать и проанализировать данные о геномных последовательностях тысяч позвоночных существ. Этой работой мы хотели сделать подарок будущим поколениям исследователей-биологов. Этот проект станет огромным скачком для всей биологии как науки и поменяет наши представления в медицине, сельском хозяйстве — во всем, что нас окружает», — рассказывает Стивен О'Брайен.

Стивен О'Брайен. Источник: siliconrepublic.com
Как продолжает профессор, такой масштабный труд стал возможен, опять же, благодаря появлению новых, более совершенных технологий, которых раньше никто не представлял. В частности, сборка ДНК-последовательностей на уровне хромосом позволила закрыть пробелы в результатах, полученных ранее.
«Когда десять лет назад мы имели на руках расшифрованные последовательности генома, это было здорово, но у них было множество ошибок и пробелов, потому что технологии в то время были несовершенны. Теперь мы знаем, как это исправить, и можем собирать непрерывные последовательности — в том порядке, в каком они закодированы в хромосомах. Это, скорее, техническое достижение, а не научное, но очень важное для работающих в этой сфере специалистов. Сегодня мы имеем гораздо более хороший доступ к информации и не сбиваемся с толку из-за ошибок в секвенировании».
Геномные данные помогут решить глобальные задачи, которые стоят перед современной функциональной генетикой: узнать, как работают ДНК-последовательности, что именно кодируют конкретные участки, какие из них являются важными, а какие вовсе не обязательны, на что влияют сбои в кодировании, как происходят генетические мутации и многое другое.
Paijmans et al., African and Asian leopards are highly differentiated at the genomic level, Current Biology (2021)/10.1016/j.cub.2021.03.084



