Как работает GATOM
Классические метаболические процессы уже довольно хорошо изучены. Самый простой пример — гликолиз, преобразование молекулы глюкозы в молочную кислоту ради получения энергии, это подробно описано в любом учебнике по биохимии. Но живые организмы далеко не всегда функционируют по заданным схемам — в клеточных процессах нередко происходят сбои и поломки, что, в частности, становится причиной развития аутоиммунных заболеваний, дегенеративных и раковых процессов. Изучение таких «поломок» особенно важно для иммунологии — зная, что именно привело к воспалению или опухоли, можно научиться на них влиять и даже останавливать.
Инструмент, разработанный учеными ИТМО, работает так: исследователь (биолог, иммунолог, медик, биоинформатик) загружает в программу данные анализа, например, опухолевой ткани. Это могут быть данные о том, какие метаболиты присутствуют в ткани и какие гены активны в том или ином процессе. Программа сравнивает исходные данные с базами KEGG и Rhea, в которых описано большинство биологических процессов в стандартном состоянии. Итоговый результат выдается в виде графа, в котором наглядно видно весь путь превращения веществ в ходе биохимических реакций. Причем алгоритм сам вычисляет, на какие метаболиты и гены обратить внимание:
«Программа раскрашивает граф так, что яркие цвета или большие размеры соответствуют более интересным реакциям. Вершины этого графа — вещества, линии между ними (ребра) соответствуют реакциям, а реакциям соответствуют определенные гены. На самом деле это достаточно нетривиальная задача — в большом графе алгоритм выделяет маленькие подграфы, в которых изменения проявлены сильнее всего. Таким образом, исследователь видит не только всю картину целиком, но и отдельные важные процессы — то, как сильно меняется конкретный ген или метаболит, как эта реакция связана с другими реакциями. Это позволяет более точно и верно интерпретировать данные, а значит, лучше понимать, что происходит, строить гипотезы, как на эти процессы можно влиять», — объясняет директор научно-образовательного центра геномного разнообразия ИТМО и руководитель фронтирной лаборатории «Вычислительные методы для системной биологии» Алексей Сергушичев.
Алексей Сергушичев. Фото: ITMO.NEWS
Благодаря разработанному вычислительному методу ученым уже удалось связать развитие болезни Альцгеймера с «поломкой» в гене TREM2. А еще — показать, что снизить темпы роста опухоли можно за счет замедления определенных процессов метаболизма в раковых клетках. И это лишь несколько примеров потенциала программы, говорят авторы работы.
Что изменилось
Сама программа была разработана достаточно давно — первый релиз состоялся еще в 2016 году. Но как признаются разработчики, хоть она и решала свою главную задачу — анализ биохимических процессов в клетках, получаемую в результате модель было не всегда просто интерпретировать. Дело в том, что раньше вершинами графа выступали вещества — метаболиты, которые могли повторять свои превращения множество раз, создавая путаницу при интерпретации. Сейчас же разработчики предлагают новый подход — в нем акцент делается не на вещества, а на их атомы, отследить которые гораздо проще:
«Мы поняли, что на самом деле удобнее рассматривать граф превращения не веществ, а атомов. Мы точно знаем, что при реакции один атом превращается в другой — и тут нет никакой неоднозначности, как в случае с веществами. Последовательность превращений одного атома всегда соответствует последовательности реакций, которые могут идти друг за другом. Это линейная последовательность, которая соответствует нашему пониманию стандартных метаболических путей. Это позволило сильно структурировать наш граф», — поясняет Алексей Сергушичев.
Ещё одно очень важное обновление функционала — возможность работать с липидомными данными. Как объясняют разработчики, липидомика — относительно новое, но активно развивающееся направление, и программ, которые умеют анализировать метаболические липидные процессы, практически нет. Однако реакции с липидами уже хорошо описаны в базе Rhea — теперь она также является частью алгоритма GATOM.
Граф процесса расщепления глюкозы (гликолиза и глюконеогенеза). Изображение предоставлено разработчиками сервиса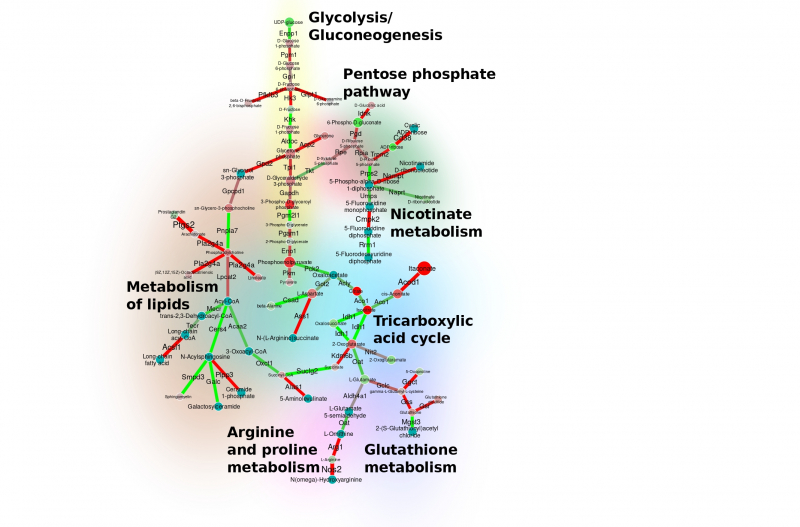
Что дальше
Раньше GATOM был доступен только как веб-сервис, но для удобства пользователей разработчики сделали скачиваемый пакет на языке R — он выложен в открытом доступе на github. Все пайплайны программы также доступны для свободного использования и модификации, чтобы пользователи могли адаптировать алгоритм под свои нужды. В оригинальной программе можно выбрать вариант только из четырех живых организмов: мыши, человека, грибка (дрожжи) и растения (arabidopsis). Но открытость исходного кода позволяет модифицировать пайплайн под любой исследуемый организм.
Сейчас алгоритм работает на основе сравнения загруженных данных со «стандартом» (чаще всего это сравнение больных клеток и здоровых), но в будущем разработчики планируют добавить возможность сравнивать сразу несколько состояний одновременно. Собственно, сам подход уже существует, но как веб-сервис он пока что работает слишком медленно (из-за увеличивающейся нагрузки на вычислительные мощности). Так что теперь задача — улучшить алгоритм и сделать его более быстрым и эффективным.
Сравнение липидомных процессов у мыши с обычной диетой и с диетой с большим количеством жиров. Изображение предоставлено разработчиками сервиса
Исследование реализовано при поддержке программы «Приоритет- 2030» в рамках фронтирной лаборатории «Вычислительные методы для системной биологии».
Статья: Mariia Emelianova, Anastasiia Gainullina, Nikolay Poperechnyi, Alexander Loboda, Maxim Artyomov, Alexey Sergushichev. Shiny GATOM: omics-based identification of regulated metabolic modules in atom transition networks (Nucleic Acids Research, 2022).



