Зачем изучать экспрессию генов
Экспрессия генов — это процесс, при котором информация, закодированная в генах, используется для создания белков и других молекул, с помощью которых клетки выполняют свои функции. Изучение экспрессии генов может помочь лучше понять механизмы различных заболеваний, так как дает знание о том, как и какие белки или молекулы вовлечены в развитие этих заболеваний.
Существует много общедоступных баз данных, в которых хранится информация об экспрессии генов — например, Gene Expression Omnibus (GEO) и ArrayExpress. В последнее время всё большую популярность приобретают данные секвенирования одиночных клеток. Обычно такие датасеты содержат множество немаркированных клеток, поэтому каждому исследователю приходится идентифицировать клетки самостоятельно. Без этой разметки данные об экспрессии генов сложно сделать доступными для большого количества исследователей. А всё потому, что они требуют не только дополнительной обработки образцов для идентификации клеточных типов, но и специфических знаний в конкретной области (например, специализации на иммунных клетках).
Источник: photogenica.ru
Что сделали ученые ИТМО
Чтобы систематизировать информацию об экспрессии генов, ученые ИТМО совместно с консорциумом ImmGen (научно-исследовательский проект, который создает базу данных экспрессии генов для иммунных клеток мышей; в нем участвуют Медицинская школа Вашингтонского университета в Сент-Луисе и Гарвардская медицинская школа) и Tabula Muris (проект по сбору данных транскриптома отдельных клеток мышиного организма, содержащий почти 50000 иммунных клеток из 20 органов и тканей) разработали новый цифровой ресурс. С его помощью ученые в интерактивном режиме могут узнать экспрессию генов фагоцитов из разных органов лабораторных мышей и решить, чем им могут быть полезны те или иные клетки, а также сформировать гипотезы о том, какими функциями они обладают. При создании ресурса ученые сразу идентифицировали все входящие в него типы клеток. Благодаря этому ресурс смогут использовать исследователи широкого профиля, так как им не придется самостоятельно идентифицировать типы клеток.
Для создания ресурса ученые использовали большие датасеты: ImmGen Open Source, ImmGen Phase 1 и Tabula Muris Senis. Образцы этих датасетов были взяты из 38 различных тканей организма лабораторных мышей. Ученые работали с тремя видами фагоцитов: макрофагами, дендритными клетками и моноцитами. Исследованные клетки ― резиденты своих тканей, то есть постоянно проживают в них и обладают функциями, адаптированными к конкретным тканям. Обычно извлечь такие клетки сложно, но исследовать их важно, потому что они могут обладать специфическими функциями — например, отвечают за развитие каких-либо заболеваний.
Набор тканей, из которых были взяты клетки для секвенрования: (А) Схематическое изображение тканей мыши, из которых были взяты образцы (отмечены разными цветами в зависимости от датасета); (B) Количество тканей, пересекающихся во всех датасетах; (C) Распределение образцов по типам клеток во всех датасетах. Источник: Cell Reports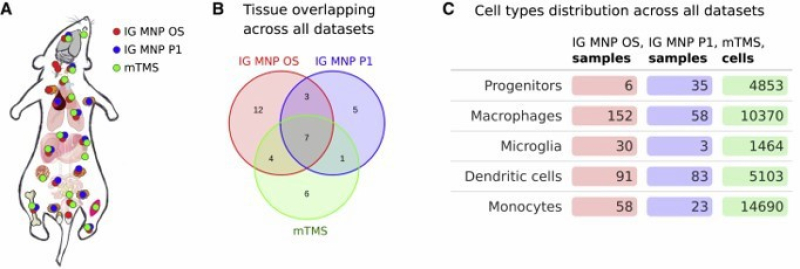
Также биоинформатики ИТМО предложили метод, который помогает описывать большие наборы данных секвенирования с метаболической точки зрения: процессов расщепления, утилизации и синтеза разных веществ в клетках. Этот метод называется GAM-кластеризация. Он позволяет находить наиболее специфичный для конкретной группы клеток связный участок в глобальной метаболической сети. Эти участки называются метаболическими модулями и содержат совместно регулируемый набор реакций, реализующий какую-то конкретную функцию в клетке.
Предложенный метод можно применить как к данным секвенирования образцов (bulk RNA-seq), так и к данным секвенирования одиночных клеток (single cell RNA-seq). Преимущество в том, что его можно использовать с неограниченно большим числом состояний клеток. При этом большинство методов в биоинформатике спроектировано для сравнения только двух типов состояний: например, только для сравнения клеток из ткани А и из ткани Б. Новый же метод позволяет использовать данные сразу для всех тканей (А, Б, В, Г и других) и найти свои метаболических особенности для каждого из состояний клеток. Раньше такую задачу не удавалось решать для данных крупного масштаба.
Ученые применили метод к данным из разработанного ими ресурса и идентифицировали основные метаболические особенности различных популяций фагоцитирующих клеток. Это позволило оценить, как меняются эти особенности в зависимости от типа клеток и их местоположения.
Некоторые из полученных метаболических модулей ученые проверили экспериментально — опыты подтвердили биологическую надежность получаемых с помощью GAM-кластеризации результатов. В частности, полученные модули показали, что вмешательство in vivo в синтез холестерина снижает миграционные способности дендритных клеток. А синтез глутатиона (внутриклеточного антиоксиданта) необходим для продукции лейкотриенов (группы липидов) перитонеальными и легочными макрофагами.
Источник: photogenica.ru
Перспективы разработки
«Ученым важно знать, чем метаболизм здоровых клеток отличается от метаболизма клеток при различных патологиях. В связи с этим воздействие на метаболизм макрофагов сегодня рассматривается как одна из терапевтических стратегий при лечении и профилактике некоторых заболеваний. Например, макрофаги играют решающую роль в развитии атеросклероза — хроническом заболевании артерий, при котором на стенках этих сосудов откладываются холестериновые бляшки. Когда макрофаги поглощают слишком много холестерина и становятся дисфункциональными, они могут способствовать образованию бляшек. Исследования показали, что воздействие на метаболизм макрофагов может помочь предотвратить развитие атеросклероза или даже обратить его вспять», — рассказала один из авторов исследования Анастасия Гайнуллина.
Анастасия Гайнуллина. Фото: ITMO.NEWS
Другой такой пример — рак, при котором макрофаги могут оказывать как стимулирующее, так и ингибирующее действие на опухоль в зависимости от их метаболизма. Модулируя метаболизм макрофагов, можно сместить их функцию в сторону противоопухолевого фенотипа — такой терапевтический подход считается довольно перспективным.
И хотя все еще нужно провести огромное число исследований, чтобы полностью понять роль метаболизма макрофагов в различных заболеваниях и разработать эффективные методы лечения, текущий прогресс уже выглядит довольно многообещающим, подчеркивает Анастасия Гайнуллина.
Исследование реализовано при поддержке программы «Приоритет–2030» в рамках фронтирной лаборатории «Вычислительные методы для системной биологии».
Статья: Anastasiia Gainullina, Denis Mogilenko, Li-Hao Huang, Helena Todorov, Vipin Narang, Ki-Wook Kim, Lim Sheau Yng, Andrew Kent, Baosen Jia, Kumba Seddu, Karen Krchma, Jun Wu, Karine Crozat, Elena Tomasello, Regine Dress, Peter See, Charlotte Scott, Sophie Gibbings, Geetika Bajpai, Jigar V. Desai, Barbara Maier, Sébastien This, Peter Wang, Stephanie Vargas Aguilar, Lucie Poupel, Sébastien Dussaud, Tyng-An Zhou, Veronique Angeli, J. Magarian Blander, Kyunghee Choi, Marc Dalod, Ivan Dzhagalov, Emmanuel L. Gautier, Claudia Jakubzick, Kory Lavine, Michail S. Lionakis, Helena Paidassi, Michael H. Sieweke, Florent Ginhoux, Martin Guilliams, Christophe Benoist, Miriam Merad, Gwendalyn J. Randolph, Alexey Sergushichev, Maxim Artyomov, ImmGen Consortium. Network analysis of large-scale ImmGen and Tabula Muris datasets highlights metabolic diversity of tissue mononuclear phagocytes (Cell Reports, 2023).