
Как показывают исследования ученых-генетиков, некоторые растения и даже животные обладают полногеномными дупликациями, то есть некоторые их гены присутствуют в нескольких копиях, более или менее сходных друг с другом. Очевидно, что у организма-предка такой дупликации не было, однако в какой-то момент в ходе эволюции она произошла и закрепилась в популяции.
«Иногда геном или его часть дуплицируется, изначально это происходит случайно, но изменение закрепляется и дальше идет в поколениях, — рассказывает ведущий научный сотрудник Университета ИТМО, лауреат программы ITMO Fellowship and Professorship Никита Алексеев. — К примеру, дуплицировалась какая-то хромосома, а полученные копии генов — это материал для создания новых функций в эволюции».
Чтобы понять процесс дупликации генома, необходимо построить так называемую эволюционную историю вида с этим эволюционным событием. Эта история позволяет проследить, что случалось с популяцией в прошлом, и обнаружить, где именно произошла дупликация и в каких условиях она закрепилась. Однако работа по воссозданию такой истории является сложной биоинформатической задачей.
Никита Алексеев. Фото: ITMO.NEWS
«Сборка геномов, а также воссоздание на их основе эволюционных историй — это совсем молодая область, — поясняет Алексеев. — Проблема в том, что у нас очень несовершенные входные данные, в них много ошибок, необходимо использовать и комбинировать данные, полученные разными методами. С развитием технологий, которые позволяют получать более качественные данные, появляется необходимость разработки новых математических моделей и алгоритмов для их анализа. Таким образом, чем больше мы знаем, тем более сложные вопросы встают перед нами. Это, если вам угодно, похоже на очень большой кроссворд, в котором вам даны задания даже не для каждого загаданного слова. При этом время от времени вам откуда-то прилетают кусочки заданий, в которых говорится о все новых словах по горизонтали и вертикали».
Линейное программирование
Пытаясь построить эволюционную историю видов с полногеномными дупликациями, ученый сталкивается с целым рядом задач, похожих по своей цели, однако совершенно разных по своей математической структуре. Чтобы эффективно их решить, необходимо прибегнуть к оптимизации. Для этого международная группа ученых, куда вошли специалисты Университета ИТМО и Университета Джорджа Вашингтона в США, предложила использовать приемы целочисленного линейного программирования, основы которого заложил советский математик и экономист, нобелевский лауреат Леонид Канторович.
«Есть класс близких по смыслу, но разных с точки зрения математики задач, — поясняет Никита Алексеев, который является соавтором исследования. — Для них мы разработали унифицированный подход, который сводится к целочисленному программированию. Это метод оптимизации, сводящий сложную проблему к набору линейных уравнений и неравенств, для решения которых существует ряд эффективных пакетов».
Процесс дупликации генома. Изображение предоставлено Никитой Алексеевым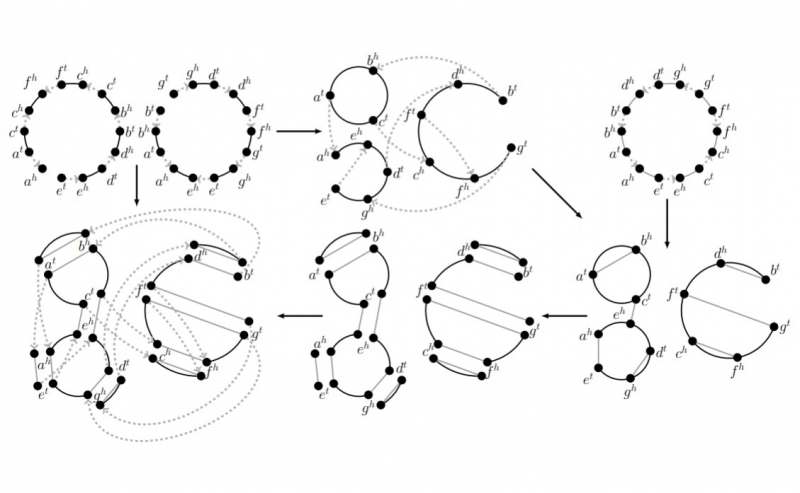
В результате ученые разработали программу, которая анализирует дуплицированные геномы и делает с высокой долей вероятности верное предположение о том, какой эволюционный путь прошел вид за свою историю, какое количество дупликаций генома произошло за это время, а также как менялись копии генов, возникшие в результате дупликации. Иногда в них самих появляются мутации, изменения определенных участков, так, что они уже не являются точными копиями друг друга.
Новый метод опробован на дрожжах, однако для масштабных биологических исследований данный метод пока не использовался. Тем не менее, авторы надеются, что их алгоритм будет пользоваться интересом у биоинформатиков разных стран. Метод может быть применим и для дуплицированных участков генома у животных.
«Дупликации участков генома встречаются у многих видов и могут затрагивать не весь геном, а только какие-то отдельные его фрагменты, и наш инструмент можно адаптировать и к таким проблемам», — заключил Алексеев.



