Не секрет, что процессы, которые протекают в нашем организме, могут быть связаны с определенными генами. Предрасположенность к облысению, полноте или плохому зрению может быть обусловлена генетически, как и некоторые болезни. Но как понять, какой именно участок генома ответственен за определенное состояние человека?
Современные технологии позволяют увидеть корреляцию между конкретным геном и процессом в организме, но зачастую это не означает, что между ними действительно есть связь. Группа ученых из Университета ИТМО предложила новый метод, который позволяет оценить вероятность существования подобной зависимости для каждого гена. Современные успехи генетики и биоинформатики позволяют лучше понимать, как работает человеческий организм, и как то, что в нем происходит, связано с генами. В СМИ то и дело появляются заголовки о том, что исследователям удалось найти «гены бессонницы», облысения или гиперактивности.
«Сравнивая гены человека, у которого есть интересующая нас болезнь или другой процесс в организме, с генами тех, у кого этой болезни нет, мы можем увидеть, активность каких генов отличается, ― рассказывает Алексей Сергушичев, доцент Университета ИТМО. ― На основе этих данных мы можем предположить, что некоторые из отличающихся генов могут иметь значение для развития интересующей нас болезни».
Однако у человека около 20 000 генов, и отличаться у исследуемых людей могут очень многие из них. Как же определить среди большого количества «подозреваемых» именно те гены, которые влияют на развитие болезни или другого состояния в организме? Ведь пока ученые не найдут именно те гены, которые связаны с интересующим их биологическим процессом, они не найдут и способ исправить их работу. При этом за болезнь отвечает не один ген, а целая группа связанных между собой участков генома ― и это тоже осложняет поиски.
Показатель важности
Сперва ученые определяют степень предполагаемой важности отличающихся по своей работе генов: она зависит от того, насколько часто и выраженно особенности в функционировании этих генов встречаются у людей с интересующим ученых диагнозом.
На основе этого ученые могут предположить, что вероятность связи одних генов с болезнью выше, чем других. Соответственно, у первых показатель важности будет больше, у вторых ― меньше. Есть также гены с нулевым и даже отрицательным показателем важности ― это значит, что исследователи либо предполагают, либо даже уверены, что эти участки генома не связаны с интересующим их заболеванием.
Когда важность каждого отдельного гена определена, ученые могут приступать к построению графа ― совокупности генов, которые могут быть связаны с заболеванием, и связей между ними. Эта задача сложнее, чем кажется, ведь нельзя просто механически объединить все гены с максимальными показателями важности в одну систему и ограничиться этим.
«Проблема в том, что какие-то гены между собой связаны биологически, а какие-то нет, соответственно, если взять все гены, которые являются важными, исходя из первичного анализа, они не будут связаны между собой, ― рассказывает Алексей Сергушичев. ― Наша задача в том, чтобы найти связанные гены. Обычно мы считаем, что между генами существует связь, если их белки, то есть продукты генов, взаимодействуют в клетке».

Но и здесь все непросто, ведь два предположительно важных гена могут быть связаны не напрямую, а иметь связь через третий ген с нулевым или даже отрицательным показателем важности, и таких комбинаций могут быть тысячи. Ни один человек не сможет вручную построить активный модуль, то есть точный граф, который бы объединил правильный набор важных генов и пути их связи. Для этого существуют сложные программные алгоритмы.
Проблема однозначных выводов
Графов, в которых найдено много предположительно важных генов и показаны пути их связи, можно построить великое множество. Однако как определить, какой же из них и есть активный модуль? Наиболее популярным подходом здесь является сравнение суммарного показателя важности. То есть, когда программа добавляет к графу ген с высоким показателем важности, то общая важность растет. Если для связи двух важных генов приходится вводить в систему ген с отрицательной важностью, то общая сумма падает. Тот вариант, у которого этот показатель окажется максимальным, и считался верным.
«В целом это работает неплохо как с точки зрения науки, так и с точки зрения прикладных применений, но есть проблема, которая давно нас беспокоила, ― поясняет Алексей Сергушичев. ― Когда мы считаем какой-то граф оптимальным, мы никак не отражаем то, каким его частям мы доверяем больше, а каким меньше. Мы включаем ген с отрицательной важностью, чтобы соединить два важных гена, но уверены ли мы, что этот ген не важен сам по себе? Кроме того, на выходе у нас получается бинарный подход ― либо ген важен, либо нет. А это не очень хорошо, ведь в разных частях графа мы все равно уверены не одинаково».

«Даже с точки зрения математики, если мы выдаем какое-то множество, которое считаем важным, а какое-то множество считаем неважным, мы, скорее всего, ошибемся хотя бы в одном месте, ― отмечает ведущий научный сотрудник Университета ИТМО, лауреат программы ITMO Fellowship and Professorship Никита Алексеев. ― Мы скажем, что какой-то ген важен, а он на самом деле не важен, мы назовем какой-то ген неважным, а это окажется не так. Вместо этого мы хотели бы предложить вероятностный подход ― показать степень нашей уверенности в важности каждого гена. Чтобы мы видели, что один ген важен с вероятностью в 35%, другой в 75%, третий в 97%».
Как построить корабль в бутылке
Чтобы решить эту проблему, группа ученых из Университета ИТМО предложила новый подход к анализу данных о вероятности связей между тем или иным процессом и генами. Вместо того, чтобы стремиться сделать один граф с наибольшей суммой важности, ученые решили построить сотни и тысячи графов с более или менее высоким показателем важности, а потом проанализировать их состав.
Однако и здесь есть математическая проблема ― как сгенерировать нужную выборку? Как сделать достаточное количество графов, которые подходят для анализа? Для этого ученые ИТМО предложили вероятностный подход ― метод Монте Карло по схеме марковских цепей (Markov chain Monte Carlo).
«Если объяснять максимально просто, представьте, что вы хотите собрать корабль в бутылке. Вы можете пытаться сделать это при помощи пинцета, а можете, очень грубо говоря, потрясти бутылку некоторое время. Когда какие-то детальки собрались так, как нам нужно, мы фиксируем систему на этом состоянии и пытаемся дальше действовать таким же методом. Если нас не устраивает, как легли детальки ― начинаем сначала, ― рассказывает Никита Алексеев. ― Рано или поздно, если мы будем достаточно долго трясти, у нас получится что-то максимально похожее на наш корабль. Это, конечно, немного потешное описание, но суть передает верно. То есть мы делаем случайные движения, и когда получается хорошо, то мы их оставляем и идем дальше, когда плохо ― забываем и пробуем снова».
Если применять эту аналогию, ученые берут какой-то набор генов и убирают из него один ― совокупная важность растет, следовательно, все сделано правильно, этот результат можно сохранить. Если дальше они убирают ген и важность падает, можно отказаться от изменения, а можно и продолжить с некоторой вероятностью: через несколько похожих шагов важность может опять резко возрасти. Таким образом программа формирует множество вариантов графов.
Получив такую выборку, ученые могут посмотреть, какие гены встречаются в ней чаще. Если какой-то ген встречается в 90% таких графов, значит исследователи могут быть уверены в его связи с исследуемым состоянием на 90%.

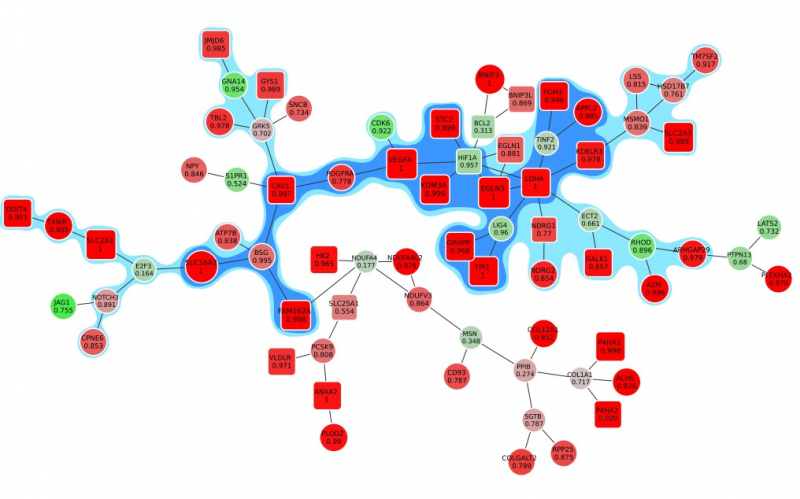
На анимации: активный модуль реакции клеток на гипоксию (недостаток кислорода). К ядру модуля постепенно добавляются вершины, в которых специалисты все менее уверены.
Проблема представления результатов
Отдельной интересной математической задачей стало то, как показать результат исследования таким образом, чтобы размер отображаемого графа зависел от допустимого уровня ошибки. Было важно, чтобы к ядру графа, где находятся наиболее вероятные «виновники» биологического процесса, по мере увеличения допустимой ошибки последовательно присоединялись новые элементы, имеющие меньшую важность. При этом граф не должен перестраиваться, гены не должны меняться местами, запутывая исследователя.
«В будущем это может быть реализовано как система с ползунком, который мы можем двигать по шкале нашей уверенности, чем меньше мы ставим степень нашей уверенности, тем больше генов нам показывается, ― объясняет Никита Алексеев. ― Если нам нужен консервативный подход, только те гены, в которых мы уверены, то мы выставляем 99%. Если нам важна более широкая картина, хотя мы знаем, что в ней будут присутствовать ошибки, мы можем выставлять 70%, 60% и так далее».
Статья: Nikita Alexeev, Javlon Isomurodov, Vladimir Sukhov, Gennady Korotkevich, Alexey Sergushichev. Markov chain Monte Carlo for active module identification problem. BMC Bioinformatics, 2020/10.1186/s12859-020-03572-9